[Drug Name]
Common Name: Imicizumab Injection
Trade Name: Suyoulile
[Manufacturer]
ChugaiPharma Manufacturing Co., Ltd
[Ingredients]
Active ingredient: Imicizumab
Imicizumab is a modified humanized IgG4 monoclonal antibody prepared in Chinese Hamster Ovary (CHO) cells through recombinant DNA technology. The antibody has a bispecific antibody structure that can bridge coagulation factor IXa and coagulation factor X. Excipients: L-arginine, L-histidine, L-aspartic acid, poloxamer 188.
[Indications]
This product is suitable for routine preventive treatment of adults and children with hemophilia A (congenital lack of coagulation factor VIII) who have inhibitors of coagulation factor VIII to prevent bleeding or reduce the frequency of bleeding.
[Specification]
30mg (1ml)/bottle
60mg (0.4ml)/bottle
105mg (0.7ml)/bottle
150mg (1ml)/bottle.
[Usage and Dosage]
Overview
This product should be used under the guidance of a doctor with experience in the treatment of hemophilia and/or bleeding diseases.
If you need to substitute other biological products, you should obtain the consent of the prescribing physician in advance.
Treatment with bypass preparations (for example: prothrombin complex, activated recombinant human coagulation factor VII) should be discontinued one day before starting treatment with this product (see [Precautions]).
Recommended dosage
The recommended dosage is 3 mg/kg once weekly (loading dose) for the first 4 weeks, followed by 1.5 mg/kg once weekly (maintenance dose), administered by subcutaneous injection.
Dose adjustment during treatment
Dosage adjustment of this product is not recommended.
Missed Dose
If the dose is not given on the scheduled day, give the dose as soon as possible before the next scheduled dose date, then resume the regular weekly dosing schedule. Do not increase the dose to make up for a missed dose.
Special Populations
1. Children
Dosage adjustment is not recommended for pediatric patients (see [Pediatric Medication]).
2. Elderly people
Dosage adjustment is not recommended for patients ≥65 years old (see [Geriatric Medication]).
3. Renal impairment and hepatic impairment
Dose adjustment is not recommended in patients with mild renal impairment or mild or moderate hepatic impairment. This product has not been studied in patients with moderate or severe renal impairment or severe hepatic impairment.
Method of administration
This product is only for subcutaneous administration and should be used under the guidance of medical professionals. After receiving adequate and standardized training on subcutaneous injection techniques and obtaining permission from a doctor, patients can inject this product themselves or have their caregivers inject it. Self-administration is not recommended for children under 7 years of age.
Each injection should be done at a different site than the previous injection (see the instructions at the end of the manual for the optional injection area). Do not inject into moles, scars, or areas of skin that are tender, bruised, red, hard, or broken. Administration of this product on the outside of the upper arm should only be done by a caregiver or healthcare provider. During the treatment of this product, it is best to choose different anatomical parts for injection of other subcutaneous injection drugs.
Special Instructions for Use, Handling and Disposal
This product is a sterile solution, contains no preservatives, does not require dilution, and can be injected directly under the skin.
This product is a colorless to light yellow solution. Visual inspection should be performed before administration to ensure that the solution does not contain particles or discoloration, otherwise it should be discarded.
This injection vial is for single use only.
A syringe, pipette needle and injection needle are required to withdraw the solution from the bottle and then administer it subcutaneously.
When the injection volume of this product solution does not exceed 1mL, a 1mL syringe should be used. When the injection volume is 1mL to 2mL of solution, a 2~3mL syringe should be used. If the solution volume corresponding to the prescribed dose of this product exceeds 2 mL, it needs to be injected in divided doses.
If the prescribed dose requires the simultaneous withdrawal of drugs from multiple vials, do not mix emicizumab injections of different concentrations.
This product does not contain any antibacterial preservatives and should be used immediately once the medicine is transferred from the vial to the syringe.
There are specific instructions for use at the end of this manual, including the selection of syringes, pipette needles and injection needles, preparation before operation, selection and preparation of injection sites, injection preparation and operation instructions, and relevant information that should be paid attention to after injection. Please refer to the instructions for use for details.
[Adverse Reactions]
Clinical Trials
Because clinical trials vary greatly, the incidence of adverse reactions observed in clinical trials of one drug cannot be directly compared with the incidence of another drug in clinical trials, and may not reflect the incidence observed in clinical practice.
The following adverse reactions (ADRs) are summarized based on pooled data from a randomized trial (HAVEN1), a single-arm trial (HAVEN2), and a dose-finding trial. A total of 189 male patients with hemophilia A received at least one dose of routine preventive treatment with emicizumab, including 94 (50%) adult patients (≥18 years old), 38 (20%) adolescent patients (≥12 to <18 years old), 55 (29%) pediatric patients (≥2 to <12 years old), and 2 (1%) infants (1 month to <2 years old).
Seven (4%) of the 189 patients in the safety population were FVIII inhibitor-negative patients from dose-finding trials. The median exposure time across all studies was 38 weeks (range: 0.8 to 177.2 weeks).
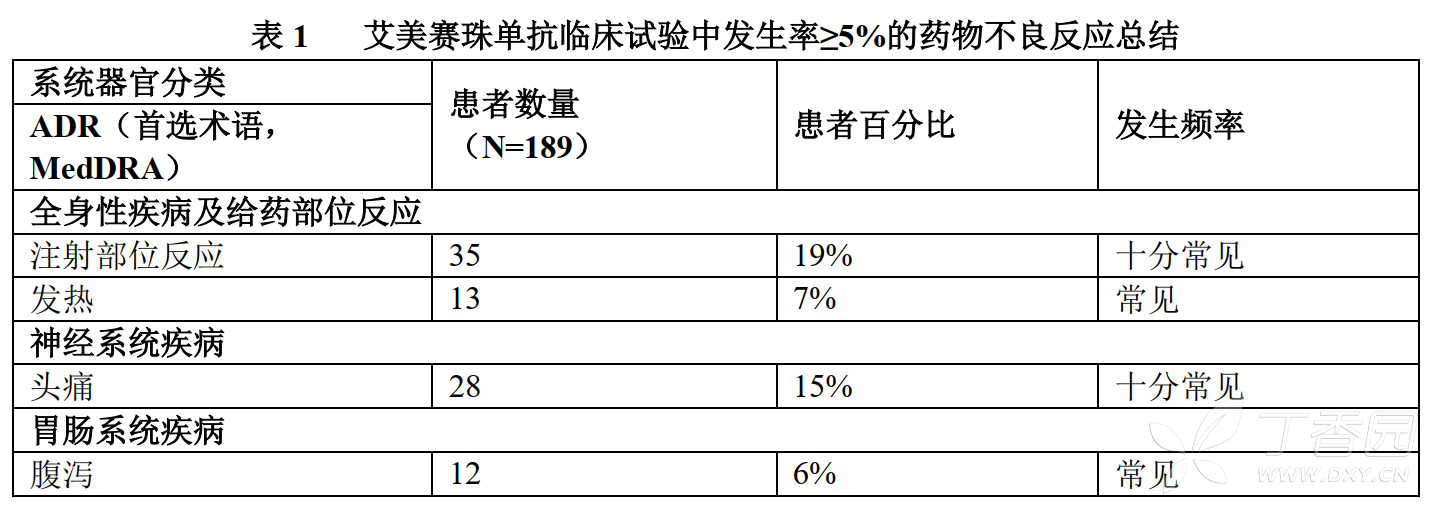
The most common ADRs observed at an incidence of ≥10% in patients who received at least one dose of emicizumab were injection site reactions, headache, and arthralgia.
In clinical trials, a total of 4 patients (2.1%) who received emicizumab prophylaxis withdrew from treatment due to ADR (thrombotic microangiopathy, skin necrosis and superficial thrombophlebitis, injection site reaction).
Table 1 below lists the adverse drug reactions that occurred in patients treated with emicizumab in clinical trials according to MedDRA system organ classification. The frequency of occurrence of each ADR is classified as follows: very common (≥1/10), common (≥1/100 to <1/10), and rare (≥1/1,000 to <1/100).
Description of special adverse drug reactions
The most serious adverse drug reactions reported in clinical trials of emicizumab were thrombotic microangiopathy (TMA) (3 cases in total) and thrombotic events (including 1 case each of cavernous sinus thrombosis and superficial venous thrombosis with skin necrosis) (see [Precautions]).
Thrombotic microangiopathy
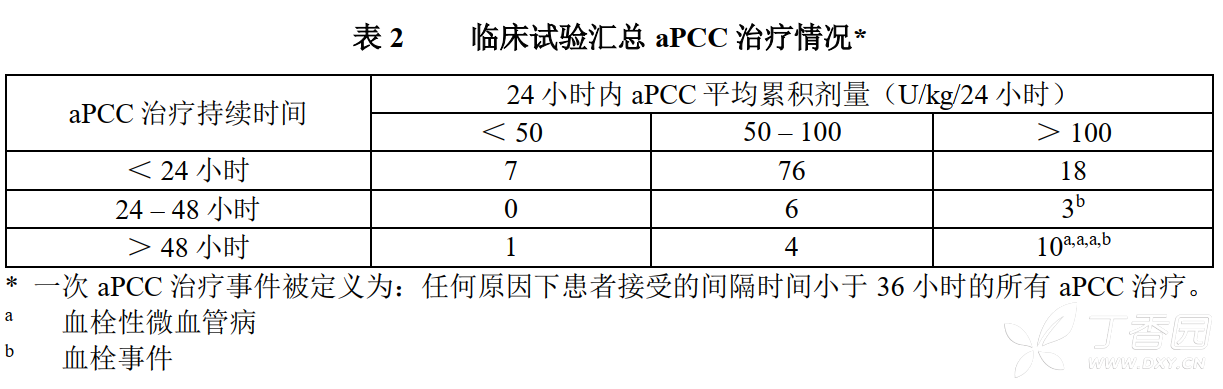
In clinical trials, the overall incidence of thrombotic microvascular events (TMA) was 1.6% (3/189) and in patients who received at least one activated prothrombin complex (aPCC) therapy during emicizumab treatment, the incidence was 8.3% (3/36). It was reported that before the occurrence of 3 TMA events (manifested by thrombocytopenia, microangiopathic hemolytic anemia, and acute renal injury without severe deficiency of ADAMTS13 activity), the patients had received aPCC treatment for 24 hours or more during emicizumab prophylaxis, and the average cumulative dose was ˃100 U/kg/24 hours. One patient restarted emicizumab treatment after remission of TMA (see Precautions).
thrombotic events
In clinical trials, the overall incidence of serious thrombotic events was 1.1% (2/189) and the incidence among patients who received at least one aPCC treatment during emicizumab treatment was 5.6% (2/36). It was reported that 2 patients had received aPCC treatment for 24 hours or more during emicizumab prophylaxis before developing thrombotic events, and the average cumulative dose was ˃100 U/kg/24 hours. One patient restarted emicizumab treatment after resolution of the event (see Precautions).
Clinical trial summary of aPCC treatment
A total of 36 patients experienced 125 aPCC treatment events, of which 13 treatment events (10.4%) in which the average cumulative dose of aPCC exceeded 100 U/kg/24 hours and lasted for 24 hours or more; among these 13 treatment events, 2 were related to thrombotic events and 3 were related to TMA (Table 2). The remaining aPCC treatment events were not related to TMA or thrombotic events.
Injection Site Reactions
A total of 35 patients (19%) reported injection site reactions (ISR). All ISRs observed in clinical trials were mild to moderate in severity, and 88% recovered without treatment. Common ISR symptoms were injection site erythema (7.4%), injection site pruritus (5.3%), and injection site pain (5.3%).
Rhabdomyolysis
Two adult patients reported rhabdomyolysis with asymptomatic elevations in serum creatine kinase without associated renal and musculoskeletal symptoms. In both patients, the events occurred after increased exercise.
Immunogenicity
As with all therapeutic proteins, treatment with emicizumab may induce an immune response. Anti-drug antibodies (ADAs), including ADAs that affect the activity of mAbs, have been detected in a small number of patients participating in clinical trials of emicizumab. The occurrence of ADA may be related to the loss of efficacy and has no obvious clinical impact on safety. No subjects in the HAVEN1 and HAVEN2 studies (n=171) were positive for anti-emicizumab antibodies. In the dose-finding study (ACE002JP), 4 subjects (n=18) tested positive for anti-Imicizumab antibodies, all of which were non-neutralizing antibodies.
The above studies used enzyme-linked immunosorbent assay (ELISA) to detect anti-Imicizumab antibodies. Immunogenicity assay results may be affected by a variety of factors, including assay sensitivity and specificity, sample handling, timing of sample collection, concomitant medications, and underlying disease. Therefore, the positive rate of anti-emicizumab antibodies may not be suitable for comparison with the positive rate of anti-drug antibodies of other products.
Post-marketing experience
No data reported.
[Contraindications]
This product is contraindicated in patients known to be allergic to emicizumab or any excipients.
[Notes]
Overview
In order to improve the traceability of biological products, the trade name and batch number of the drug should be recorded (or indicated) in the patient file. When administering this product outside of a healthcare setting, patients/caregivers are advised to record the drug lot number.
With Imicizumab and (Activation) Prothrombin complex-related thrombotic microangiopathies
In clinical trials, thrombotic microangiopathic events (TMA, see clinical trials under [Adverse Reactions]) were observed in patients who received activated prothrombin complex (aPCC) for 24 hours or more at a mean cumulative dose of ˃100 U/kg/24 hours while receiving prophylaxis with emicizumab. Treatment of TMA events includes supportive care with or without plasma exchange and hemodialysis. Improvement in TMA was observed one week after discontinuation of aPCC administration, which was different from the common clinical course observed in atypical hemolytic uremic syndrome and typical TMA (eg, thrombotic thrombocytopenic purpura) (see Clinical Trials under Adverse Reactions).
There is no clinical experience with prothrombin complex (PCC) therapy during prophylaxis with emicizumab. Based on the mechanisms of action of the two and the clinical experience of combining emicizumab with aPCC, it is speculated that PCC treatment during emicizumab prophylaxis will also increase the risk of thrombotic microangiopathy in subjects.
For patients receiving emicizumab prophylaxis, the occurrence of TMA should be monitored when aPCC/PCC is administered concurrently. If clinical symptoms and/or laboratory test results suggest the occurrence of TMA, physicians should immediately terminate the use of aPCC/PCC, suspend emicizumab treatment, and handle treatment according to the clinical situation. Physicians and patients/caregivers should weigh the benefits and risks of restarting emicizumab prophylaxis after complete remission of TMA based on the patient's individual circumstances.
Receiving aPCC or PCC treatment during emicizumab prophylaxis should be avoided if possible. If this cannot be avoided, please see the dosage recommendations for bypass agent administration below. If patients have high risk factors for TMA (e.g., previous or family history of TMA) or are receiving concomitant medications known to be risk factors for TMA (e.g., cyclosporine, quinine, tacrolimus), they should be treated with caution when receiving prophylactic treatment with this product.
Thromboembolic events associated with Imicizumab and (activated) prothrombin complex
In clinical trials, thromboembolic events were observed in patients who received Imicizumab prophylaxis concurrently with aPCC for 24 hours or longer at an average cumulative dose of ˃100 U/kg/24 hours (see Clinical Trials under [Adverse Reactions]). Anticoagulation was not required in any case, unlike conventional treatment of thrombotic events. After discontinuation of aPCC treatment, thrombotic events improved or alleviated (see clinical trials under [Adverse Reactions]).
There is no clinical experience in receiving PCC treatment during Imicizumab prophylaxis. It is speculated that PCC treatment during Imicizumab prophylaxis will also increase the risk of thromboembolic events in subjects.
For patients receiving emicizumab prophylaxis, the occurrence of thromboembolism should be monitored when aPCC/PCC is administered concomitantly.
If clinical symptoms, imaging findings and/or laboratory test results suggest the occurrence of a thrombotic event, doctors should immediately terminate the use of aPCC/PCC, suspend emicizumab treatment, and handle the situation according to the clinical situation. Physicians and patients/caregivers should weigh the benefits and risks of restarting emicizumab prophylaxis after complete resolution of the thrombotic event based on the individual patient's circumstances.
Receiving aPCC or PCC treatment during emicizumab prophylaxis should be avoided if possible. If this cannot be avoided, please see the dosage recommendations for bypass agent administration below.
Guidelines for the use of bypass agents in patients receiving emicizumab prophylaxis
Treatment with bypass agents should be discontinued one day before starting emicizumab therapy.
Before receiving prophylactic treatment with emicizumab, patients should be fully informed of the possibilities and risks of concomitant use of bypass agents. If use of a bypass agent concurrently with emicizumab prophylaxis is necessary, the physician should discuss the exact dosage and frequency of administration of the bypass agent to be used with the patient and/or caregiver.
Imicizumab can increase the coagulation function of patients. Therefore, the dose of the bypass agent required may be lower than that without emicizumab prophylaxis. The dose and duration of treatment with bypass agents also depend on the site and extent of bleeding, as well as the patient's clinical status.
Activated prothrombin complex (aPCC): aPCC should be avoided unless there are no other treatment options. If patients receiving emicizumab prophylaxis require aPCC treatment, the starting dose should not exceed 50 U/kg, and strict laboratory monitoring (including but not limited to renal function, platelet count, and thrombosis-related tests) should be performed. If bleeding cannot be controlled with a starting dose of 50 U/kg aPCC, additional doses of aPCC should be given under medical guidance or supervision, but the total aPCC dose within the first 24 hours of treatment should not exceed 100 U/kg. If a PCC dose higher than the maximum dose of 100 U/kg is required during the first 24 hours of treatment, the attending physician must carefully weigh the TMA, thromboembolic risk, and bleeding risk.
Prothrombin complex (PCC): There is currently no clinical experience with the combination of Imicizumab and PCC in clinical trials and post-marketing settings, and patients receiving Imicizumab should avoid concurrent PCC treatment. If PCC is the only treatment option for the patient, he should be treated at a specialized hemophilia treatment center and closely monitored for the occurrence of thromboembolic events or thrombotic microangiopathy.
Activated recombinant human FVII (rFVIIa): In clinical trials, patients receiving emicizumab prophylaxis only used rFVIIa, and no TMA or thrombotic events were observed.
After discontinuing emicizumab prophylaxis, dosing guidelines for bypass agents should be followed for at least 6 months (see Pharmacokinetics, Clearances).
Affects laboratory coagulation test results
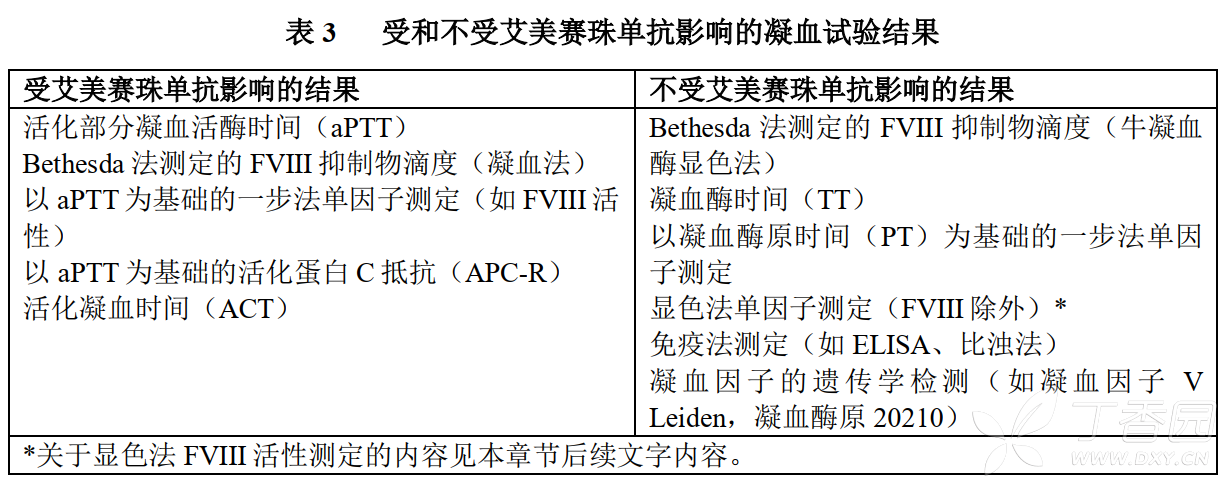
Imicizumab can restore the missing tenase cofactor activity of activated coagulation factor VIII (FVIIIa), thus affecting endogenous coagulation-based coagulation Pathway laboratory tests include activated coagulation time (ACT), activated partial thromboplastin time (aPTT) measurement results, and all aPTT-based measurement results, such as the one-step coagulation factor VIII activity measurement (see Table 3 below).
Therefore, patients receiving emicizumab cannot rely on laboratory test results based on the intrinsic coagulation pathway to monitor emicizumab activity, determine coagulation factor replacement therapy or anticoagulant dosage, or determine factor VIII inhibitor titers. Laboratory tests that are and are not affected by Imicizumab are shown in Table 3 below.
The chromogenic method can be used to determine FVIII activity using human or bovine coagulation protein. Assay methods containing human coagulation factors are affected by Imicizumab and may overestimate the clinical hemostatic effect of Imicizumab. In contrast, assays containing bovine coagulation factors are insensitive to emicizumab (no activity detected) and can be used to monitor the activity of endogenous or infused exogenous FVIII, or to determine anti-FVIII inhibitor titers.
Imicizumab is active in the presence of FVIII inhibitors and will therefore produce false negative results when measuring FVIII inhibitory function using the Bethesda coagulation assay. Therefore, FVIII activity can be measured using the Bethesda chromogenic method in bovine serum, which is insensitive to emicizumab.
Due to the long half-life of emicizumab, the above effects on coagulation test results may last up to 6 months after the last dose (see [Pharmacokinetics], Clearance).
[Drug Interactions]
Adequate or well-controlled drug interaction studies have not been conducted with Imicizumab.
Clinical experience shows that there is a drug interaction between emicizumab and aPCC (see clinical trials under [Precautions] and [Adverse Reactions]).
Preclinical experiments suggest that coadministration of emicizumab with rFVIIa or FVIII may cause blood hypercoagulability. Imicizumab enhances coagulation, so the dose of coagulation factors required to treat bleeding may be lower than that without emicizumab for prophylaxis.
[Packaging]
1 bottle/box
[Drug classification]
Enzymes, other therapeutic drugs

.jpeg)